Services
Services
- Developmental delays: crawling, walking
- Hand clapping or hand biting
- Hyperactive or impulsive behavior
- Mental retardation
- Speech and language delay
- Tendency to avoid eye contact
- Physical signs: Flat feet, flexible joints and low muscle tone, large body size, large forehead or ears with a prominent jaw, long face, large testicles, soft skin
The FMR1 KO Mouse Model
The lack of FMRP in these mice not only affects the physical structure of neurons but also significantly alters synaptic plasticity. This alteration manifests as impaired long-term potentiation in the cortex and hippocampus, alongside an increased long-term depression in both the hippocampus and cerebellum. Such changes in synaptic behavior are essential for understanding the learning and memory deficits associated with Fragile X syndrome. Male FMR1 knockout (KO) mice, particularly those bred on an FVB/n background at PsychoGenics, are extensively utilized in research studies. They offer a valuable tool for exploring the mechanisms of Fragile X syndrome and developing potential therapeutic strategies to mitigate its effects.
Body Weight

Locomotor Activity

Rearing Activity
Rearing Activity: Fmr1 KO mice show increased rearing activity in the open field test at 10 and 14 weeks of age.

Time Spent in Center of Open Field

Electrophysiology and Biomarkers
BDNF Levels


Long-Term Depression
Long-Term Depression: Fmr1 KO mice exhibit enhanced hippocampal mGluR-dependent long-term potentiation (LTD), which is reversed by mGluR antagonist 8-OH-DPAT.
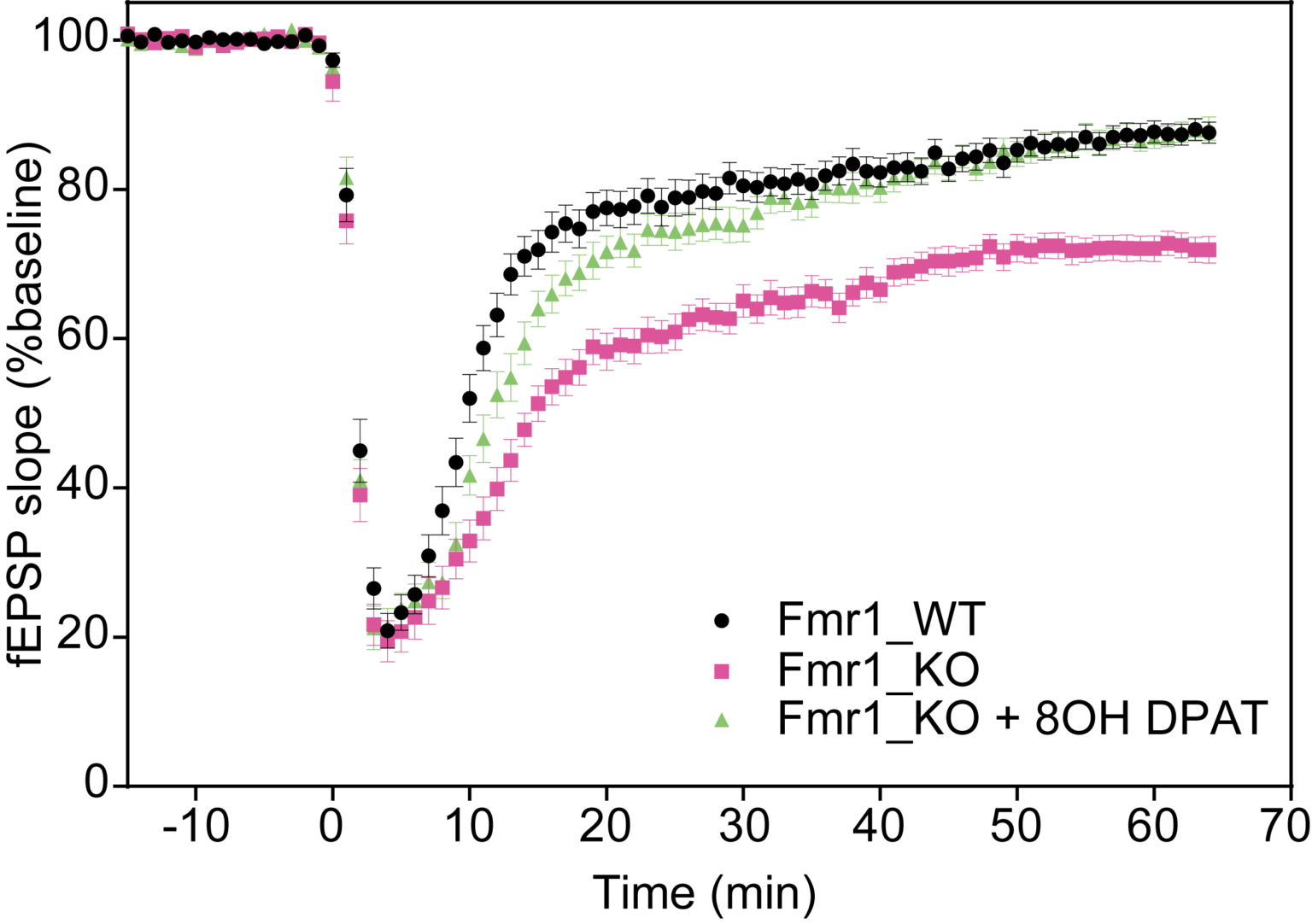
Time course of the changes in responses recorded in CA1 area of hippocampus induced with an application of an mGluR agonist (S)-DHPG (100μM).
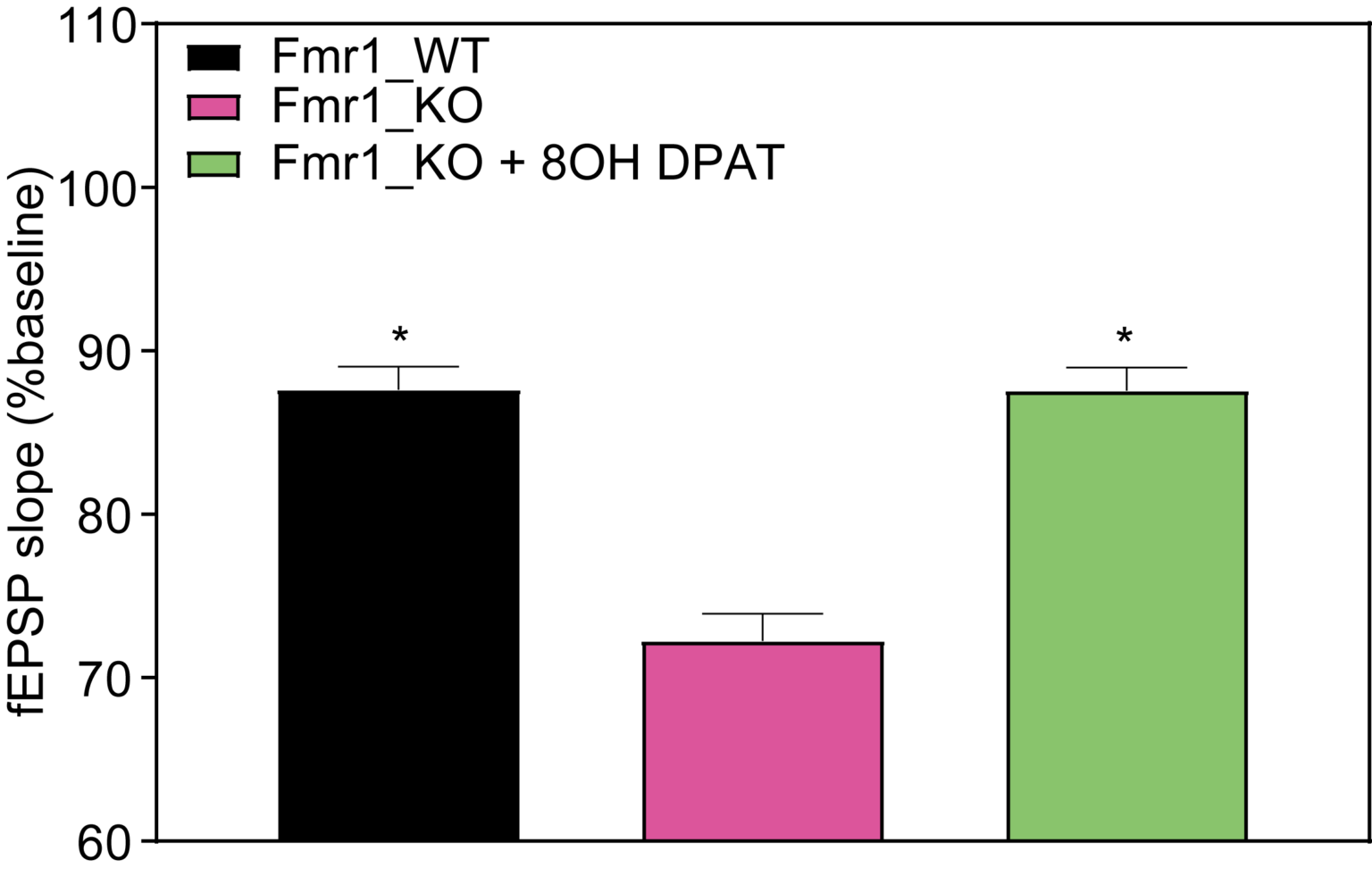
Summary of the data for the last 5 min of experiment. *p<0.05 compared to Fmr1 KO
Reduced Tonic Inhibition in Dentate Gyrus
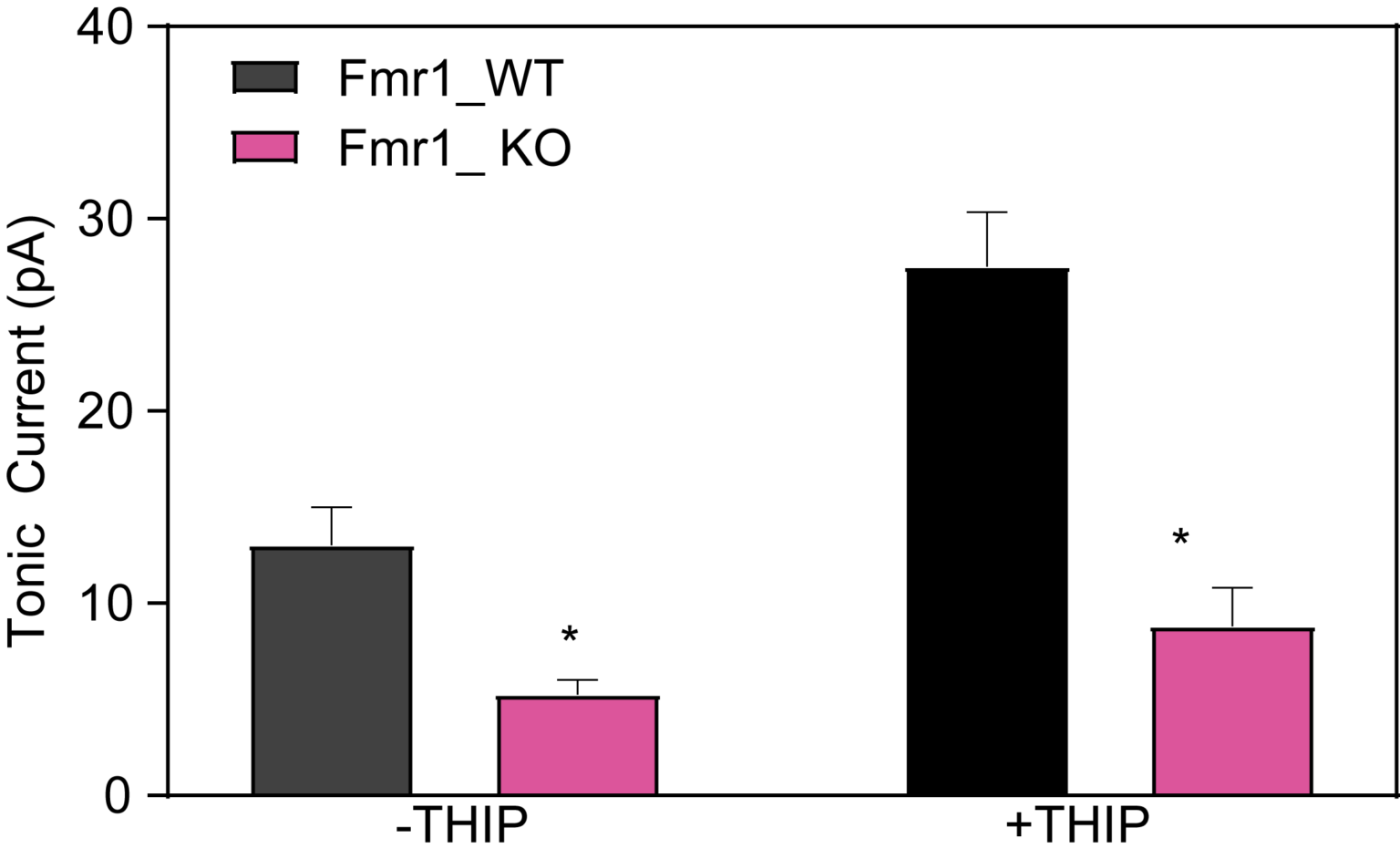
Following a stable baseline and subsequent enhancement of tonic GABA currents by the d subunit-selective agonist THIP (gaboxadol, 1 mM; +THIP), tonic currents were unmasked by blocking GABAA receptors with 100 mM picrotoxin.
Enhancement of tonic current by application of THIP normalized to baseline