Rett syndrome is a neurodevelopmental disorder caused by mutations in the X-linked methyl CpG binding protein 2 (Mecp2) gene. Primarily affecting girls, it stands as a leading cause of intellectual disabilities. Preclinical studies of Rett syndrome are pivotal for advancing treatment development. Our team brings extensive preclinical research experience to support you in discovering the next therapeutic breakthrough for Rett syndrome.
Mecp2 Mouse Model of Rett Syndrome mode of Rett Syndrome
Mice engineered with a knockout allele of the fragile X mental retardation syndrome 1 gene (Fmr1) on the X chromosome serve as a critical model for studying Fragile X syndrome, mirroring many of the phenotypic characteristics observed in humans with the condition, such as hyperactivity, repetitive behavior, and seizures. This genetic modification results in the absence of the Fragile X mental retardation protein (FMRP), a change that triggers the activation of the RAC1 protein. The consequence of this activation is a series of abnormalities in dendritic spines across various brain regions, leading to altered synaptic function. These alterations are pivotal in understanding the neurobiological underpinnings of Fragile X syndrome, as they provide insight into the disorder’s impact on neuronal architecture and connectivity.
The lack of FMRP in these mice not only affects the physical structure of neurons but also significantly alters synaptic plasticity. This alteration manifests as impaired long-term potentiation in the cortex and hippocampus, alongside an increased long-term depression in both the hippocampus and cerebellum. Such changes in synaptic behavior are essential for understanding the learning and memory deficits associated with Fragile X syndrome. Male FMR1 knockout (KO) mice, particularly those bred on an FVB/n background at PsychoGenics, are extensively utilized in research studies. They offer a valuable tool for exploring the mechanisms of Fragile X syndrome and developing potential therapeutic strategies to mitigate its effects.
Female HET Mice Show Deficits in Rotarod Performance.
Mice are placed on the continuous rotating rod (Columbus Rotamex Treadmill Apparatus) and are given a 5-min training period at a slow speed (4 RPM). After a 1 h-ITI, mice are exposed to three 5 min-trials.
- During each trial, the mice are placed back on the rotarod, and the speed is gradually and uniformly increased (0-40 RPM).
- The time that each mouse remains on the rotating rod before falling as well as the speed of the rotarod when the mouse falls is recorded.
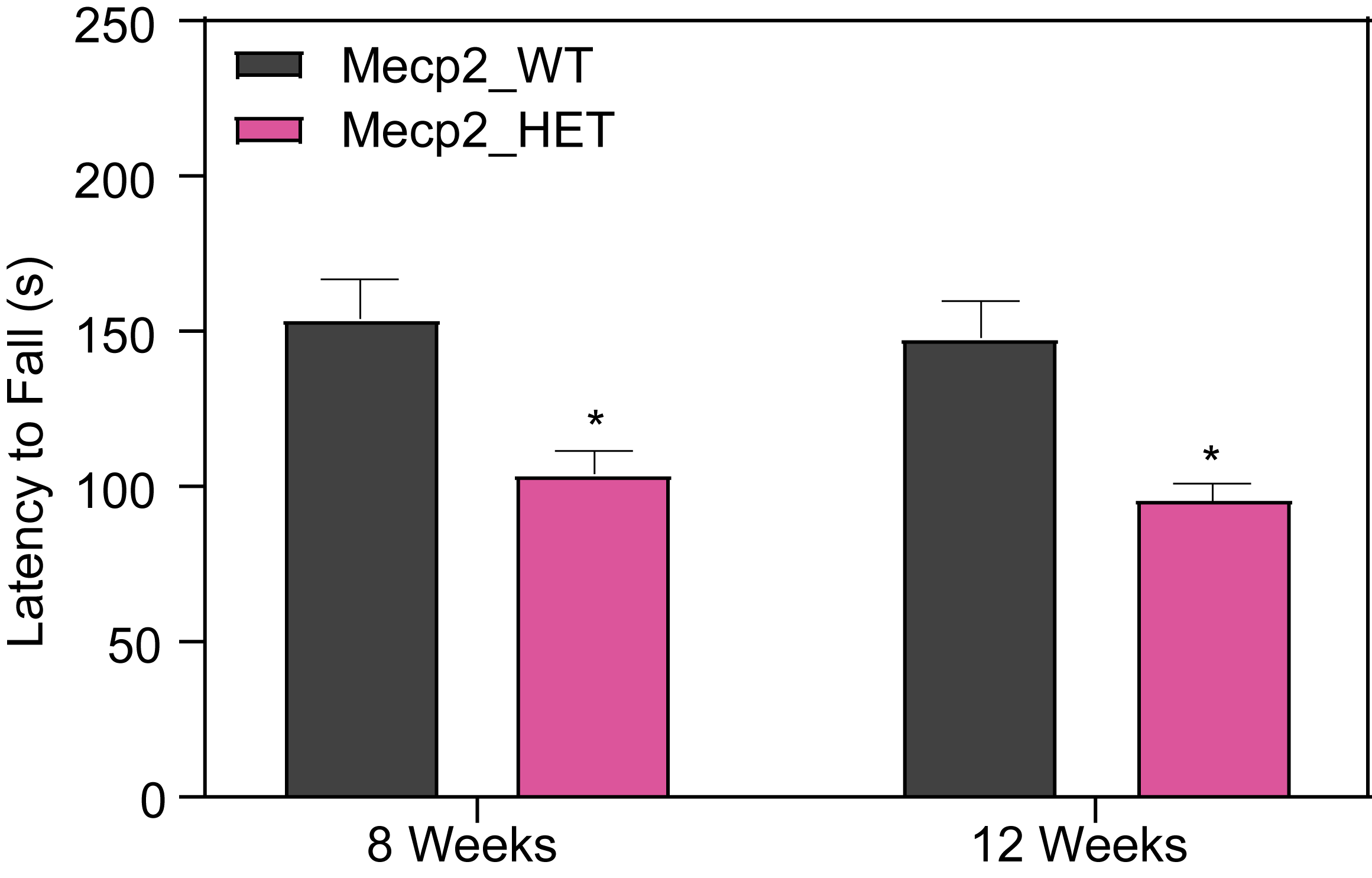
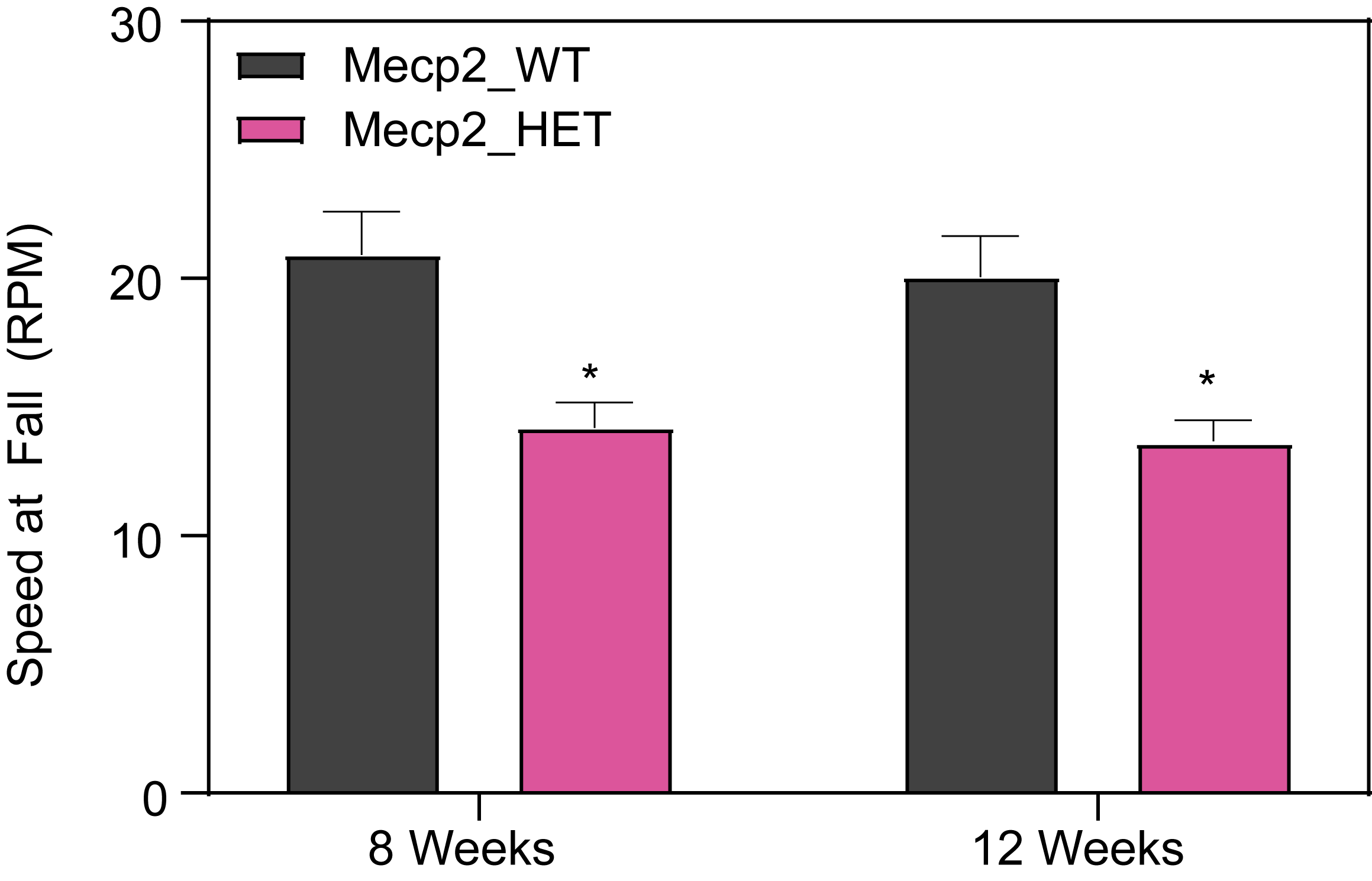
Female HET mice show increased hindlimb clasping.
- Mice are lifted gently by the tail with front limbs remaining on surface.
- Normal response is a spread in the hind limbs.
- The percent of mice that show hindlimb clasping is recorded.
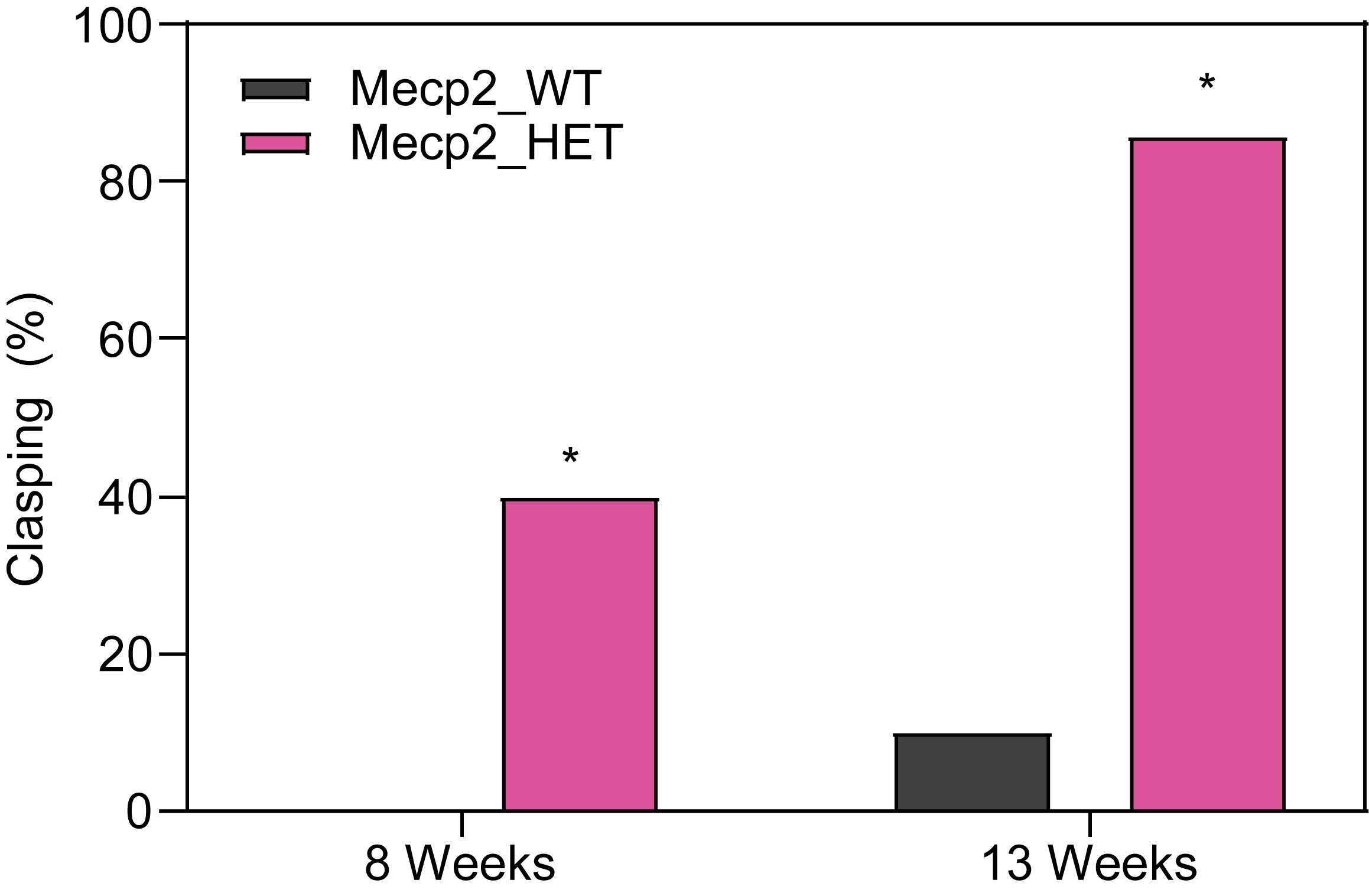
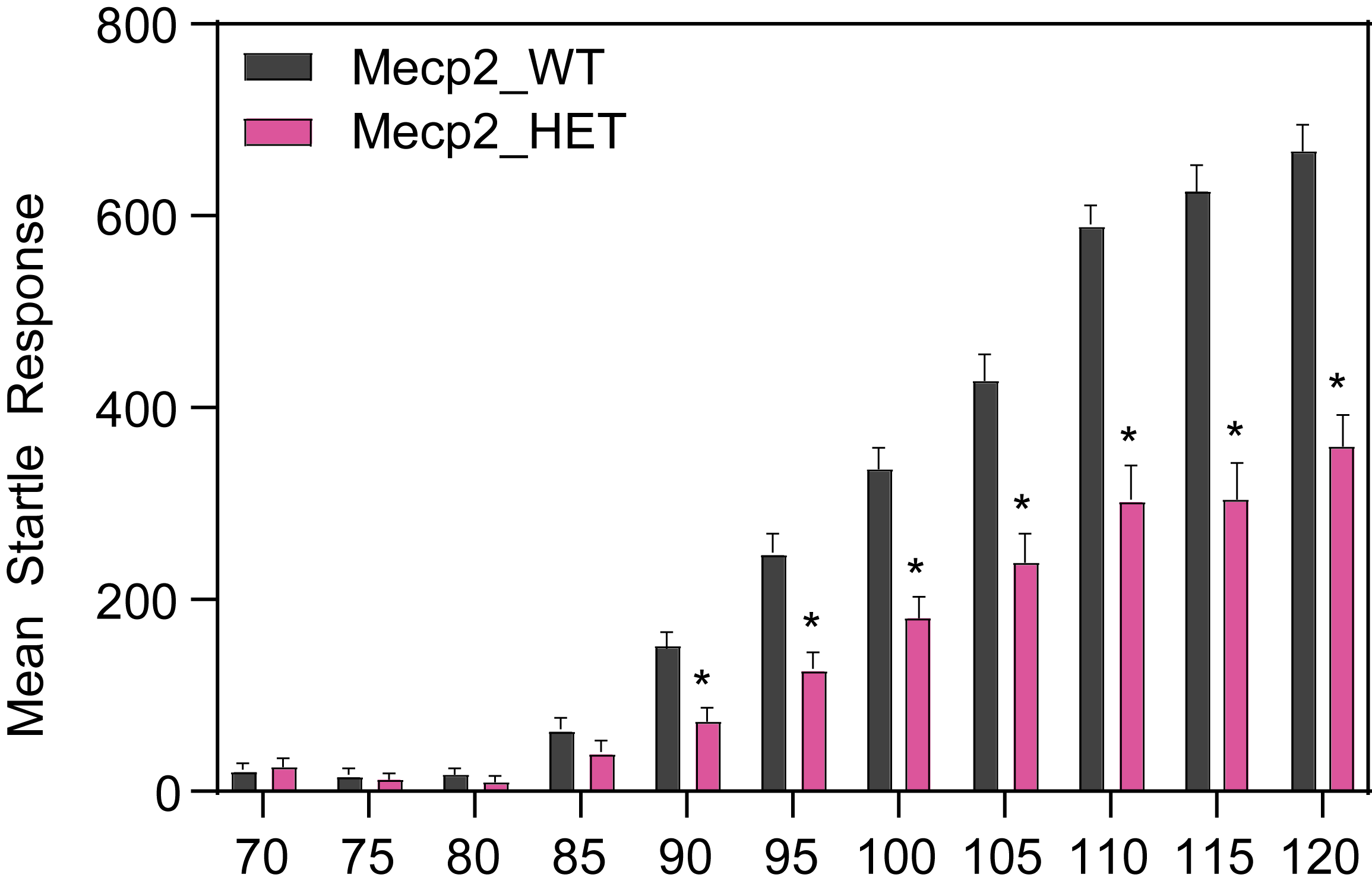
Female HET mice show decreased startle response.
- The acoustic startle measures an unconditioned reflex response to external auditory stimulation.
- Mice are placed in the startle enclosures for a 5min habituation period of white noise (70 dB).
- Subsequent test sessions consist of 10 blocks of eleven trials each. Within each block, stimuli of 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, and 120dB are presented in random order with a variable inter-trial interval of mean 15 sec (10-20sec).
- The duration of the stimulus is 40 ms. Responses are recorded for 150 ms from startle onset and are sampled every ms
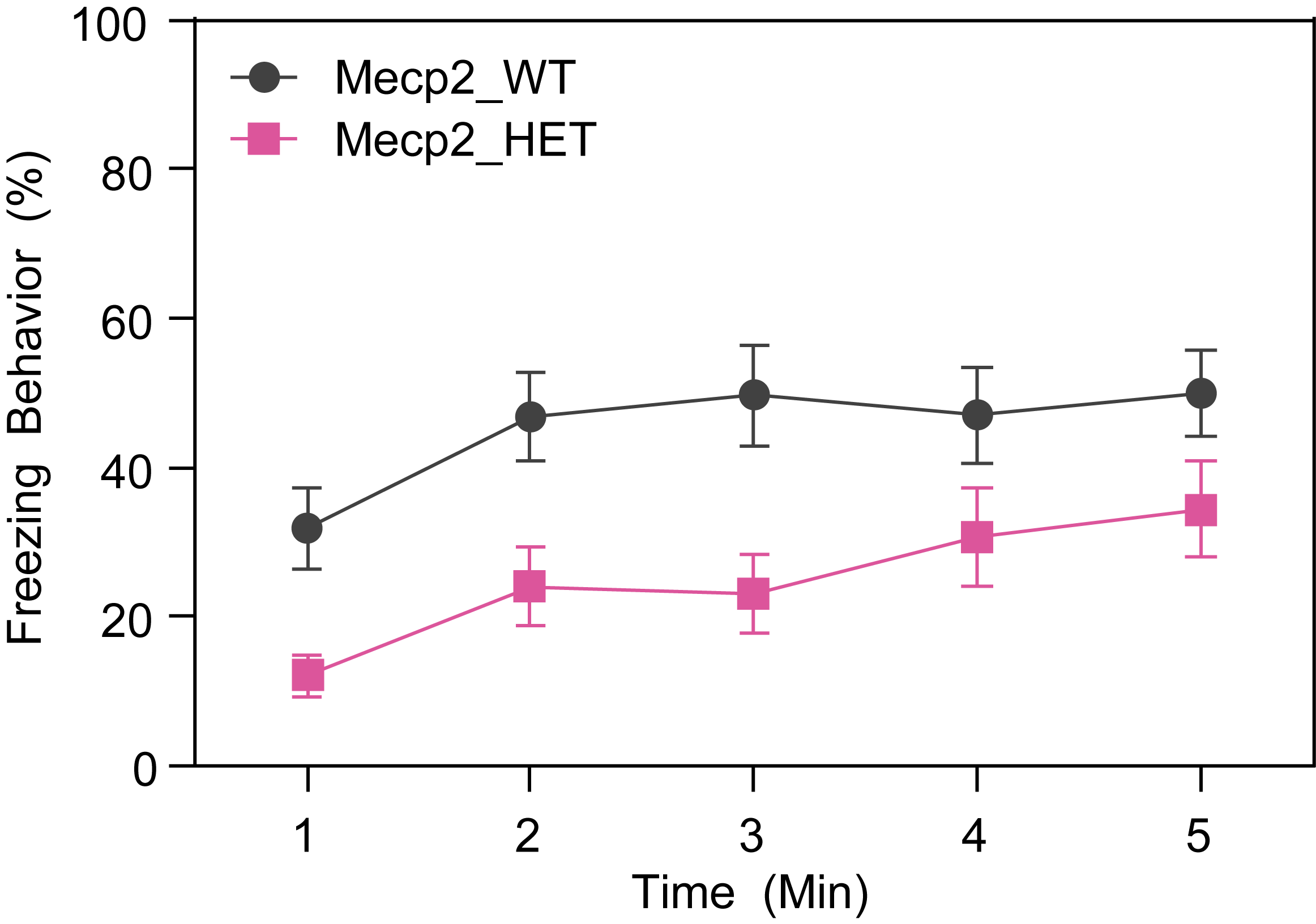
Female HET mice show decreased freezing during contextual fear conditioning test at 13 weeks of age.
- On day 1, mice are placed in a conditioning chamber to habituate to the context for 2 min (CS). A tone will be presented for 20 sec followed by a foot shock (1s, 0.5 mA; US) . This pairing of the CS and US is repeated for a total of 3 times, with an interval of 60 sec between pairings. Mice will remain in the conditioning chamber for another 60 s and then be returned to its home cage.
- On the second day, animals are tested for contextual memory in the same chamber they were trained in for a period of 5 min without shock or any other interference. Freezing is measured by FreezeView software.
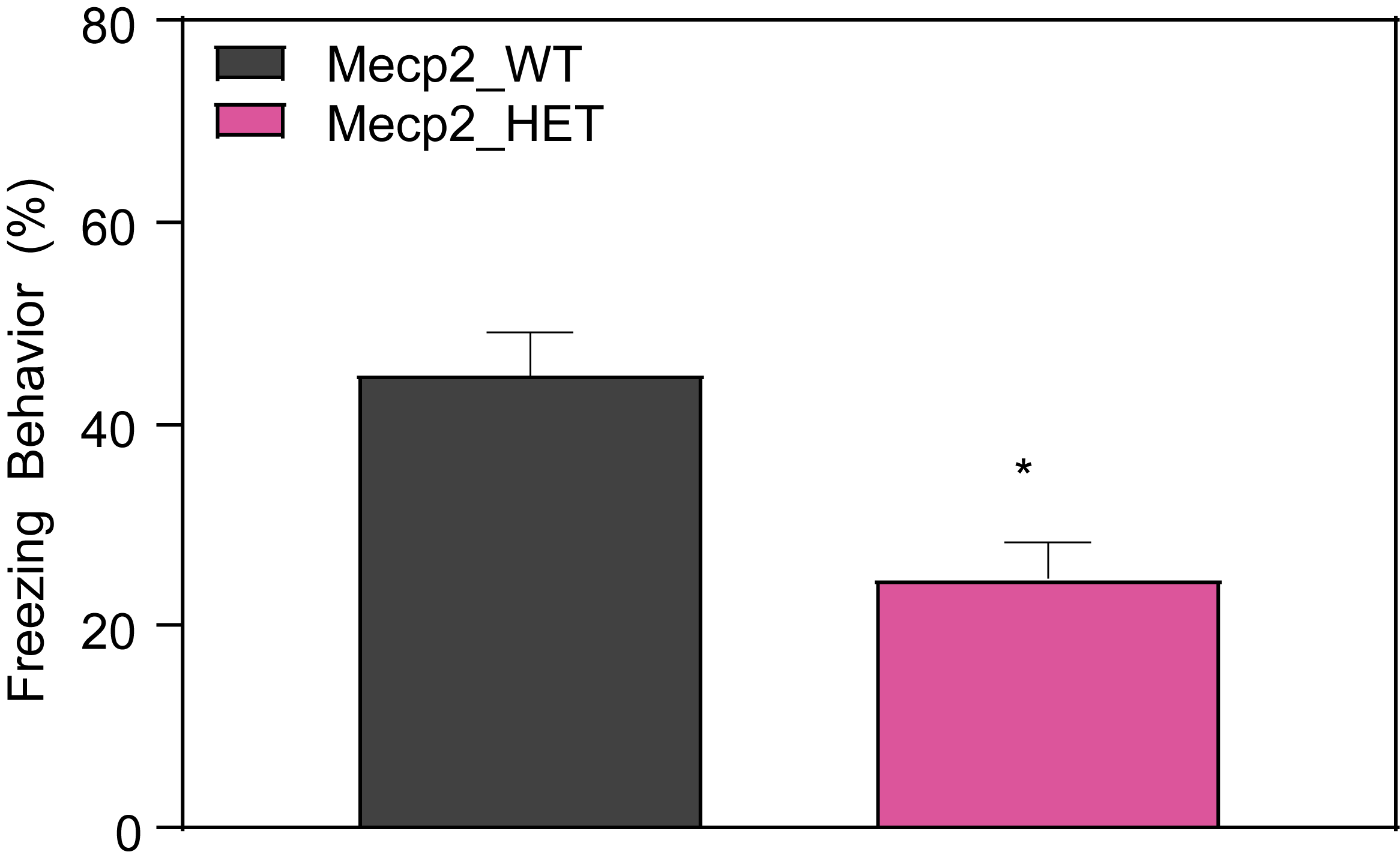
Respiration and Optokinetic Response (6-7 Month Old Mice)
Female HET mice show higher apnea counts and higher breathing rate.
- Mice are tested in a whole-body plethysmograph system for 1 hour
- Mice are awake during testing
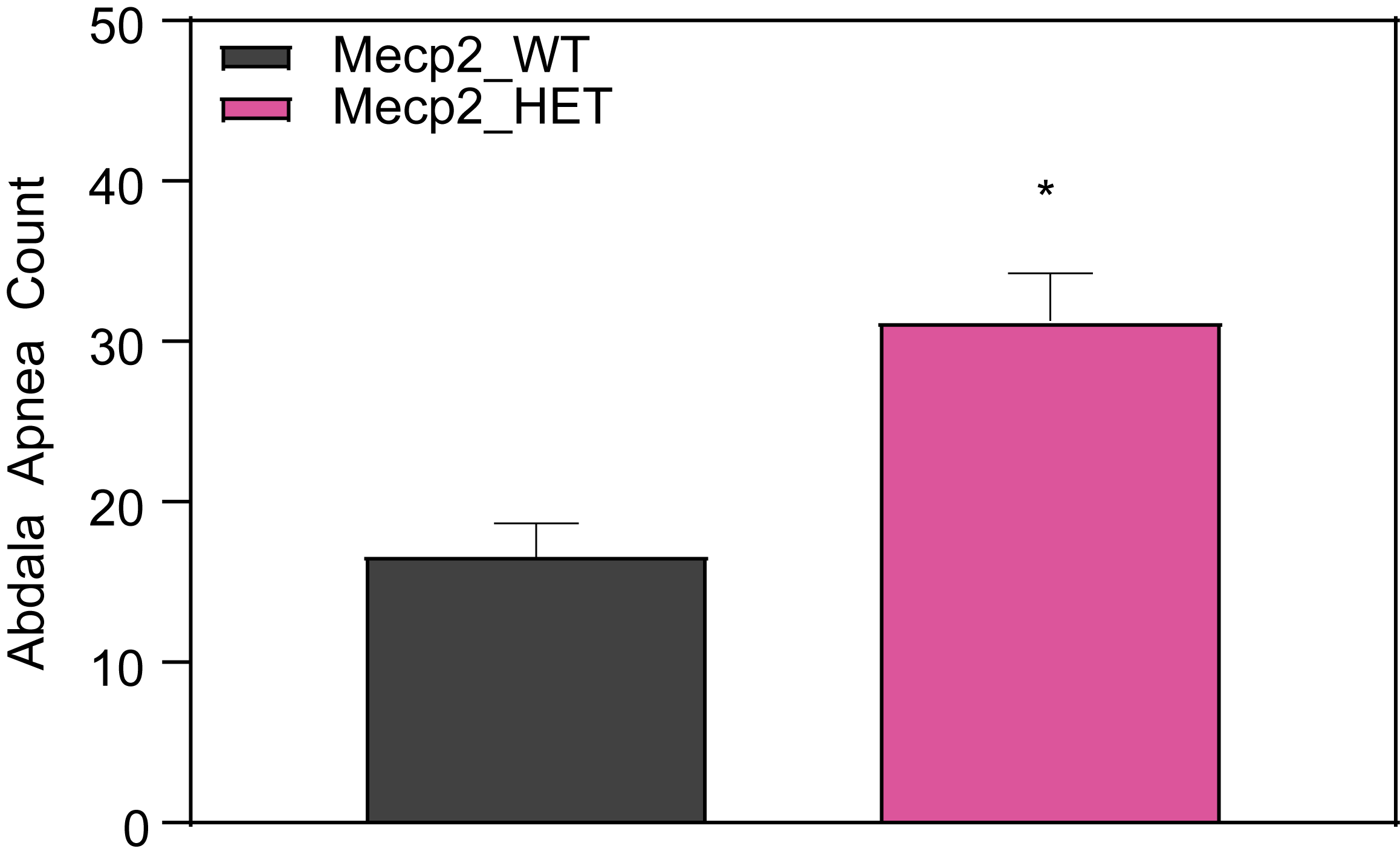
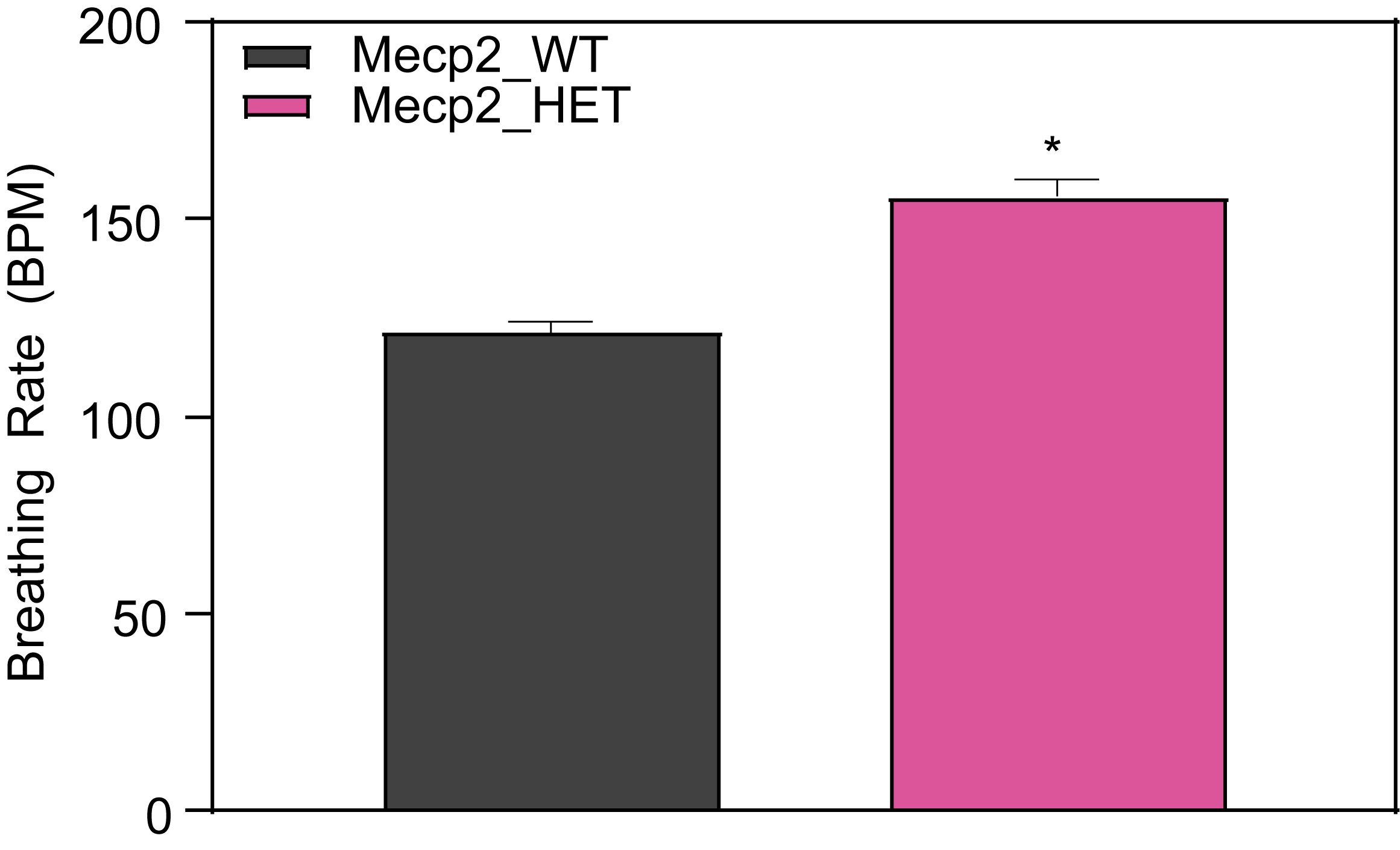
Female HET mice make more errors when tracking a moving object.
- The optokinetic response and bar tracking are scored by an observer blind to the animal strain, bar rotation, and frequency.
- Visual performance when tracking a moving object is impaired in the HET mice resulting in fewer correct optokinetic responses compared to WT mice.
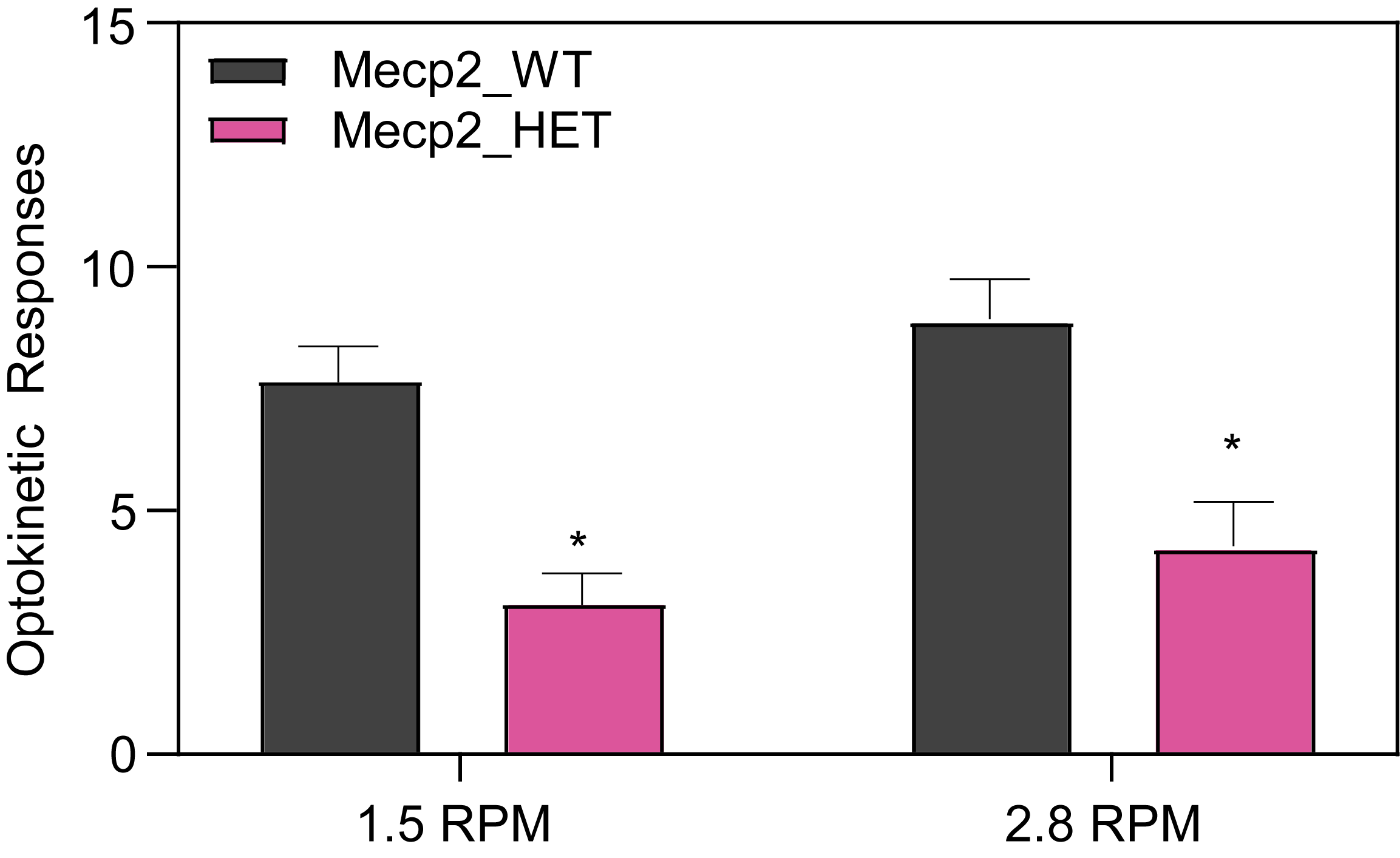
Molecular Biomarkers and Electrophysiology
Hippocampal levels of various BDNF isoforms is decreased in female HET mice.
- BDNF genes are direct molecular targets of MeCP2 and are decreased in the HET mice compared to WT mice
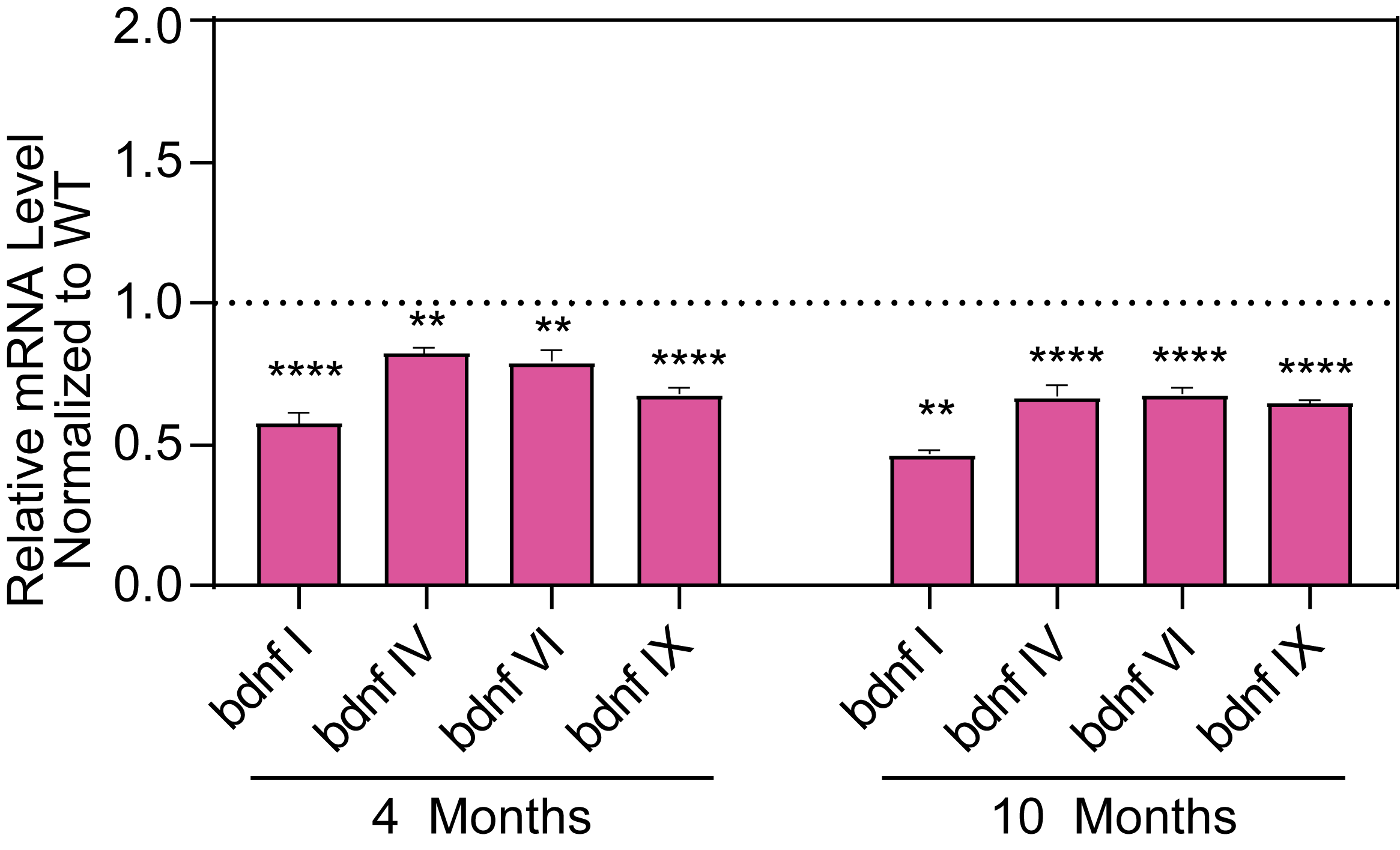
MeCP2 mice show decreased levels of PSD95, Synaptophysin Sapap3, Kir4.1 and increased levels of glutamate receptor subunits including GluR1, GluR2, NR2A, and NR2B.
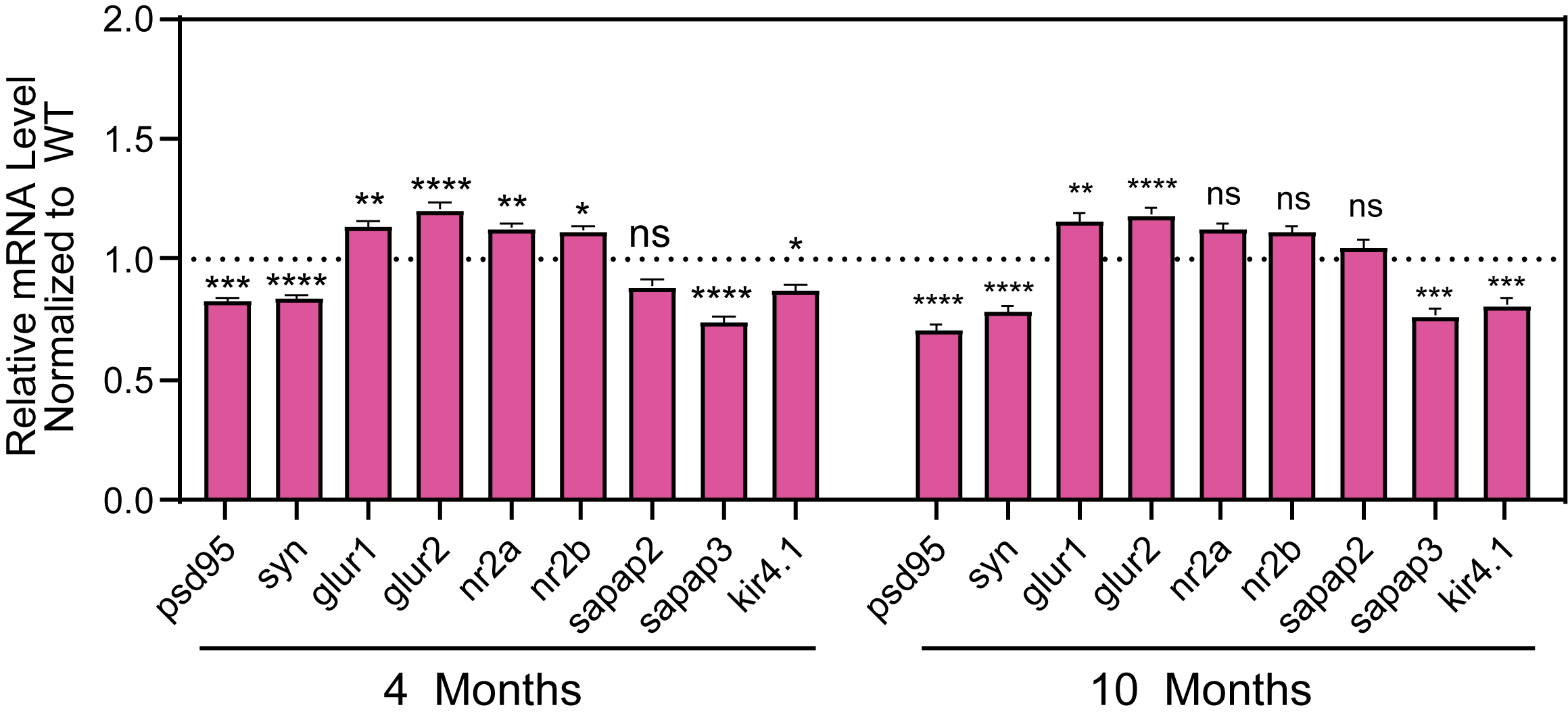
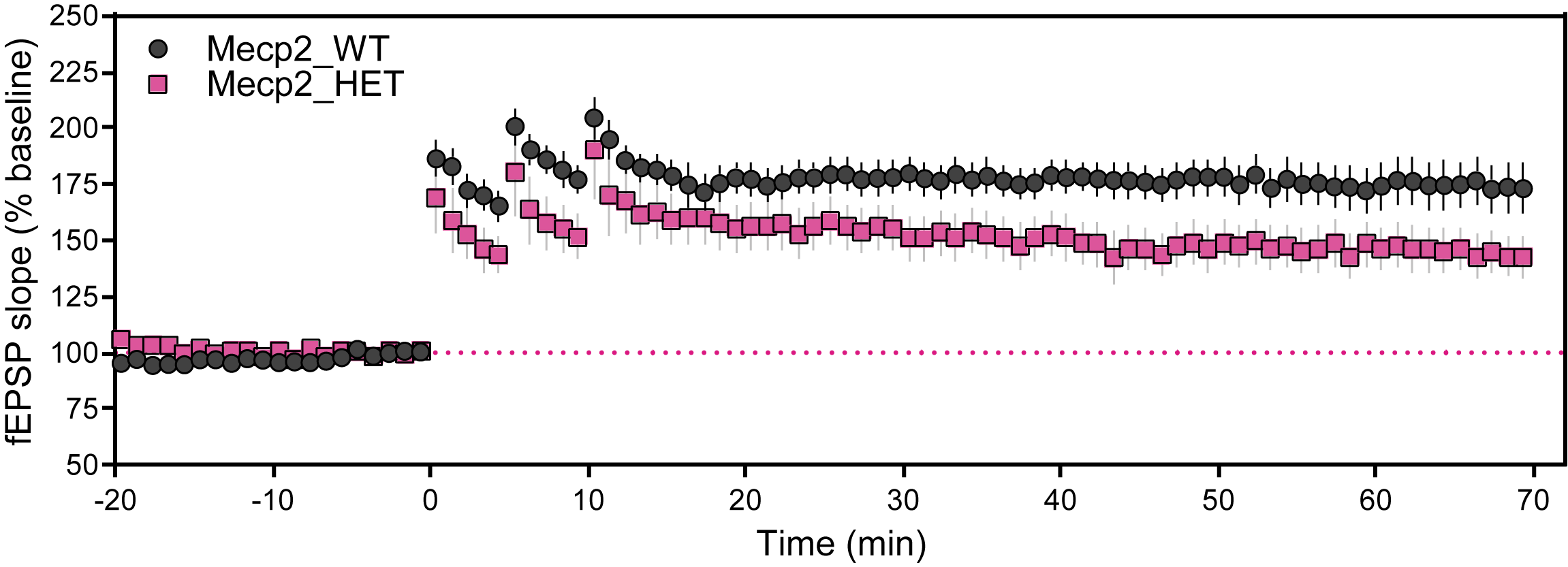
- Extracellular field excitatory postsynaptic potentials (fEPSP) were evoked in hippocampal slices at the Schaffer collateral-CA1 synapse from 6-month-old female WT and MeCP2 mice.
- Stable baseline recordings were obtained with fEPSPs evoked using 40% of maximum stimulus intensity. LTP was induced by delivering three trains of 100 Hz high frequency stimulation, each train separated by five minutes inter-train interval.
- Synaptic responses were normalized and expressed as percent from baseline.
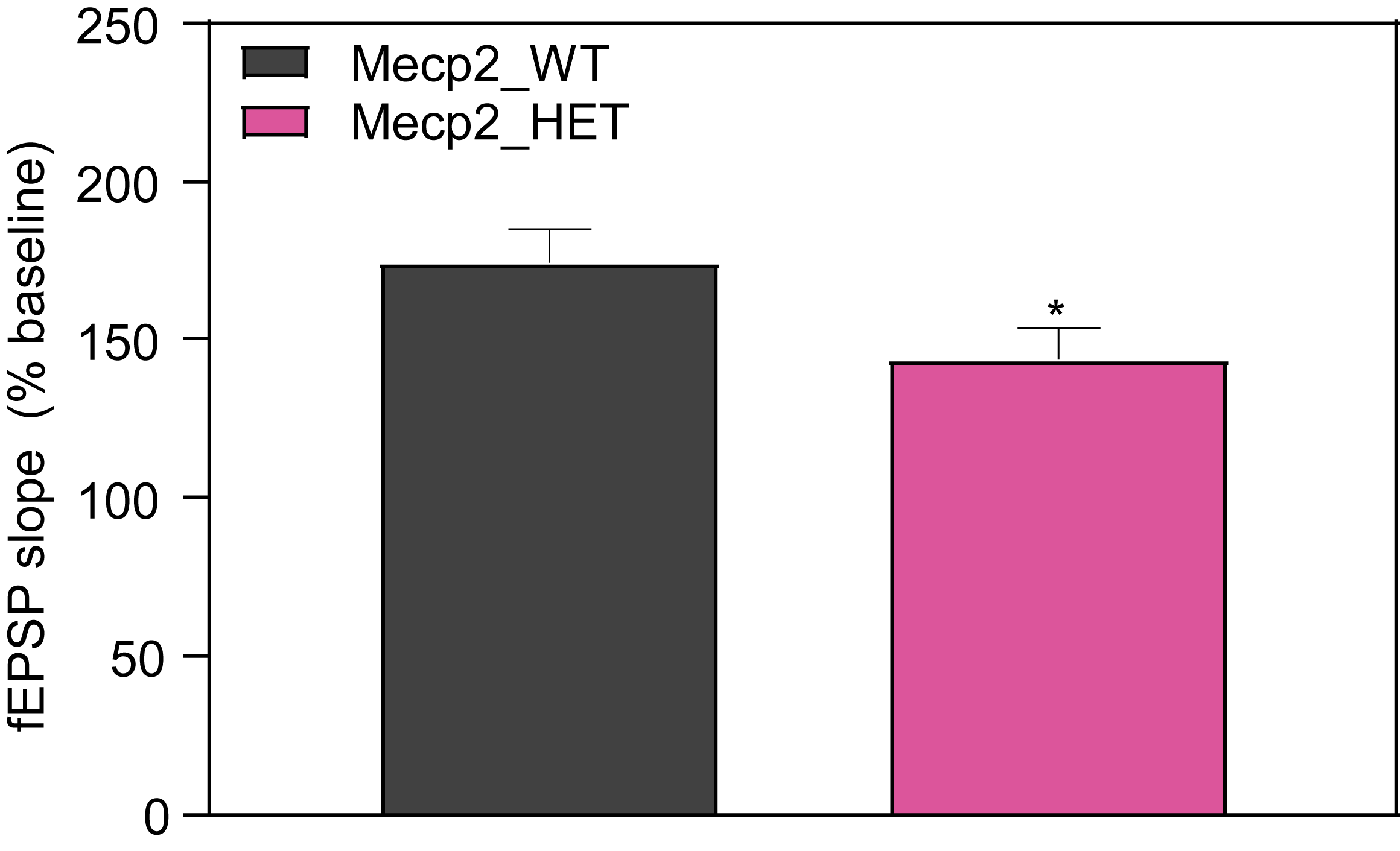
Driving Your Preclinical Rett Syndrome Research Forward
At PsychoGenics, we collaborate closely with the leading researchers in Rett syndrome research, harnessing the power of the Mecp2 heterozygous knockout mouse model to explore the underlying mechanisms of Rett syndrome and give you the best opportunity to discover your breakthrough.
Our neurodevelopmental disorders areas of expertise: